Dr Hudan Liu’s group at Frontier Science Center for Immunology and Metabolism, Medical Research Institute, Wuhan University, reports a previously unsuspected mechanism of MYC activation promoting T-cell leukemogenesis, which was recently published in Cancer Cell(2020, 37 (2), 200-215).
Dysregulation of MYC occurs in almost 50% of human cancers. The unleashed MYC oncogene frequently produces abundant MYC protein, which mediates a transcriptional response involved in a variety of biological processes, contributing to almost every aspect of tumorigenesis. The significance of MYC deregulation hasbeen documented in T-cell acute lymphoblastic leukemia (T-ALL), an aggressive hematological malignancy accounting for 15% of pediatric and 25% adult acute lymphoblastic leukemia. Current clinical treatment of T-ALL involves implementation of intensive chemotherapies which results in unneglectable toxicities, particularly to young patients. Target therapies with greater efficacy and lower toxicities are in desperate need. It has been well-established that MYC acts as an oncogenic driver in T-cell leukemogenesis and particularly in maintaining leukemia-initiating cell activity. Inhibition of MYC therefore could be a powerful approach for the treatment of T-ALL. However direct targeting of MYC has remained unsuccessful owning to its “undruggable” protein structure, and indirect inhibition of MYC has been emerging as an alternative strategy.
To identify associated factors that sustain MYC stability and transcriptional activity, Liu and colleagues performed a mass spectrometry in MYC-overexpressing Jurkat cells and identified Aurora kinase B (AURKB) as a specific MYC binding partner. Mechanistically, AURKB directly phosphorylates MYC at serine 67 (S67), counteracting the GSK3β-directed threonine 58 (T58) phosphorylation and subsequent FBXW7-mediated proteasomal degradation. The S67 phosphorylation-mediated MYC stabilization is independent of the canonical S62 phosphorylation, which is a well-characterized modification conferring to MYC stabilization and also priming for T58 phosphorylation. Studies in zebrafish T-ALL demonstrate that the S67 phosphorylation of MYC significantly contributes to MYC stabilization and T-cell leukemogenesis in vivo. As such, pharmacological inhibition of AURKB by AZD1152 abrogates MYC S67 phosphorylation, induces prominent MYC degradation and eradicates tumor growth in vitroand in vivo. Notably, inactivating FBXW7mutations, occurring in about 20% of T-ALL cases, predict resistance to AZD1152-based therapies due to the incapability of these T-ALL cells to elicit efficient MYC degradation. This study not only sheds light on an unconventional mode of MYC phosphorylation-mediated protein stabilization, but also provides a mechanism-based therapeutic strategy to target MYC and treat T-ALL. These data propel an immediate clinical trial of AURKB inhibitors in treatment of T-ALL, or perhaps other MYC-dependent cancers with FBXW7mutational status as a biomarker.
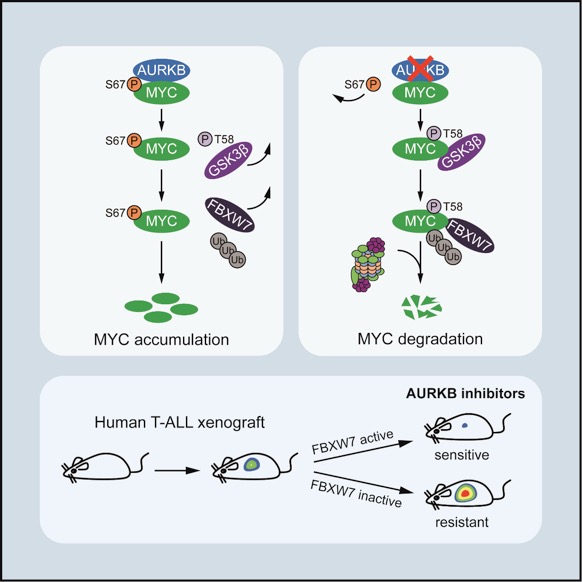
FigureAURKB induces MYC S67 phosphorylation and promotes MYC protein stability. AURKB inhibition elicits MYC degradation and shows in vivoefficacy in xenograft models of human T-ALL cells expressing active FBXW7.